Small variants evaluation
Author: Andrea Guarracino
Synopsis
The major histocompatibility complex (MHC) is a large locus in vertebrate DNA containing a set of closely linked polymorphic genes that code for cell surface proteins essential for the adaptive immune system. In humans, the MHC region occurs on chromosome 6.
Steps
Data collection
We map HPRC chromosome 6 contigs to the MHC ALT haplotypes from GRCh38:
wfmash -t 16 -m -s 5000 -p 98 MHC.fa.gz chr6.pan.fa > HPRCy1.pan.chr6_vs_MHC.paf
Then, we make a BED file for the mappings and merge the regions in it to make a single BED record per matching contig:
awk '{ print $1, $3, $4 }' HPRCy1.pan.chr6_vs_MHC.paf | tr ' ' '\t' | sort -V > HPRCy1.MHC.bed
bedtools merge -d 1000000 -i HPRCy1.MHC.bed > HPRCy1.MHC.merge.bed
Finally, we extract the FASTA file for the regions:
bedtools getfasta -fi /lizardfs/erikg/HPRC/year1/parts/chr6.pan+refs.fa -bed HPRCy1.MHC.merge.bed | bgzip -c -@ 16 > HPRCy1.MHC.fa.gz
samtools faidx HPRCy1.MHC.fa.gz
Pangenome Sequence Naming
Contigs already respect the Pangenome Sequence Naming specification (PanSN-spec).
cut -f 1 HPRCy1.MHC.fa.gz.fai | head -n 6
HG00438#1#JAHBCB010000040.1:22870000-27725000
HG00438#2#JAHBCA010000042.1:22875000-27895000
HG00621#1#JAHBCD010000020.1:22865000-27905000
HG00621#2#JAHBCC010000005.1:28460000-33400000
HG00673#1#JAHBBZ010000030.1:28480000-33310000
HG00673#2#JAHBBY010000018.1:0-2900000
Divergence estimation
The human MHC locus presents a high sequence divergence, so we will set the mapping identity (-p parameter) in pggb to 95.
Sequence partitioning
As the dataset is small, we do not need to partition sequences (see the Sequence partitioning tutorial for more information).
Pangenome graph building
To build the MHC pangenome graph, execute:
pggb -i HPRCy1.MHC.fa.gz -p 95 -n 90 -t 16 -G 13117,13219 -o HPRCy1.MHC.s10k.p95.output
Graph statistics
To collect basic graph statistics, execute:
odgi stats -i HPRCy1.MHC.s10k.p95.output/*.final.gfa -t 16 -S
#length nodes edges paths
5315371 309186 429323 126
Identify variants with vg
To call variants for each contig, execute:
vg deconstruct -P chm13 -H '?' -e -a -t 48 HPRCy1.MHC.s10k.p95.output/HPRCy1.MHC.fa.gz.39ffa23.e34d4cd.be6be64.smooth.final.gfa | \
bgzip -c -@ 16 > HPRCy1.MHC.s10k.p95.output/HPRCy1.MHC.fa.gz.39ffa23.e34d4cd.be6be64.smooth.final.chm13.vcf.gz
-H '?' avoids managing the path name hierarchy when calling variants, then emitting variants for each contig.
To filter variants (by using nesting information from the pangenome graph) and realign reference and alternate alleles, execute:
vcfbub -l 0 -a 100000 --input HPRCy1.MHC.s10k.p95.output/HPRCy1.MHC.fa.gz.39ffa23.e34d4cd.be6be64.smooth.final.chm13.vcf.gz | \
vcfwave -I 1000 -t 48 | bgzip -@ 16 \
> HPRCy1.MHC.s10k.p95.output/HPRCy1.MHC.fa.gz.39ffa23.e34d4cd.be6be64.smooth.final.chm13.vcfbub.a100k.wave.vcf.gz
Take SNPs from the PGGB VCF file:
REF=chm13#chr6:28380000-33300000.fa
NAMEREF=chm13
cut -f 1 HPRCy1.MHC.fa.gz.fai | grep chm13 -v | while read CONTIG; do
echo $CONTIG
bash vcf_preprocess.sh \
HPRCy1.MHC.s10k.p95.output/*.vcfbub.a100k.wave.vcf.gz \
$CONTIG \
1 \
$REF
done
Identify variants with nucmer
Prepare the reference FASTA file:
samtools faidx HPRCy1.MHC.fa.gz chm13#chr6:28380000-33300000 > chm13#chr6:28380000-33300000.fa
Align each sequence against the reference with nucmer:
REF=chm13#chr6:28380000-33300000.fa
NAMEREF=chm13
mkdir -p nucmer/
cut -f 1 HPRCy1.MHC.fa.gz.fai | grep chm13 -v | while read CONTIG; do
echo $CONTIG
PREFIX=nucmer/${CONTIG}_vs_${NAMEREF}
samtools faidx HPRCy1.MHC.fa.gz $CONTIG > $CONTIG.fa
nucmer $REF $CONTIG.fa --prefix "$PREFIX"
show-snps -THC "$PREFIX".delta > "$PREFIX".var.txt
rm $CONTIG.fa
done
Using the nucmer2vcf.R script, generate VCF files for each sequence with respect to the reference with nucmer:
REF=chm13#chr6:28380000-33300000.fa
NAMEREF=chm13
NUCMER_VERSION="4.0.0beta2"
cut -f 1 HPRCy1.MHC.fa.gz.fai | grep chm13 -v | while read CONTIG; do
echo $CONTIG
PREFIX=nucmer/${CONTIG}_vs_${NAMEREF}
Rscript nucmer2vcf.R "$PREFIX".var.txt $CONTIG $REF $NUCMER_VERSION $PREFIX.vcf
bgzip -@ 16 $PREFIX.vcf
tabix $PREFIX.vcf.gz
done
Variants evaluation
Prepare the reference in SDF format for variant evaluation with rtg vcfeval:
rtg format -o chm13#chr6:28380000-33300000.sdf chm13#chr6:28380000-33300000.fa
Compare nucmer-based SNPs with PGGB-based SNPs:
REFSDF=chm13#chr6:28380000-33300000.sdf
NAMEREF=chm13
cut -f 1 HPRCy1.MHC.fa.gz.fai | grep chm13 -v | while read CONTIG; do
echo $CONTIG
PREFIX=nucmer/${CONTIG}_vs_${NAMEREF}
PATH_PGGB_VCF=HPRCy1.MHC.s10k.p95.output/HPRCy1.MHC.fa.gz.*.smooth.final.chm13.vcfbub.a100k.wave.${CONTIG}.max1.vcf.gz
# Merge regions closer than 1000 bps to define the callable regions where to evaluate the variants
dist=1000
rtg vcfeval \
-t $REFSDF \
-b $PREFIX.vcf.gz \
-c $PATH_PGGB_VCF \
-T 16 \
-e <(bedtools intersect -a <(bedtools merge -d $dist -i $PREFIX.vcf.gz ) -b <(bedtools merge -d $dist -i $PATH_PGGB_VCF)) \
-o vcfeval/${CONTIG}
done
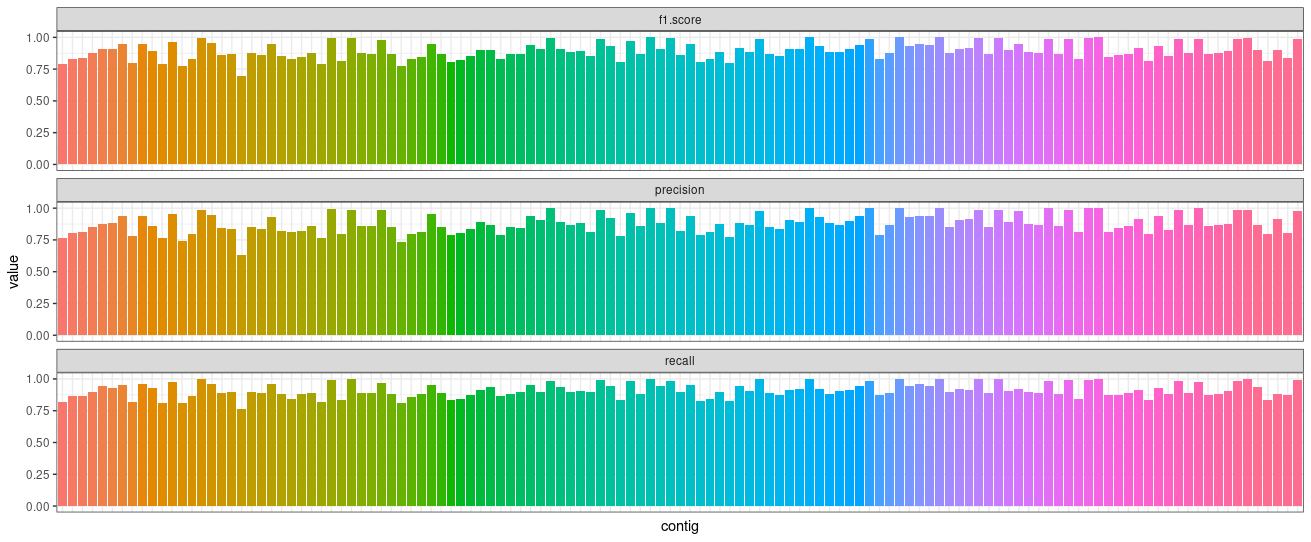
Collect statistics:
cd vcfeval
(echo contig precision recall f1.score; grep None */*txt | sed 's,/summary.txt:,,' | tr -s ' ' | cut -f 1,7,8,9 -d ' ' ) | tr ' ' '\t' > statistics.tsv
Plot statistics:
require(ggplot2)
require(tidyr)
stat_df <- read.table('statistics.tsv', sep = '\t', header = T, comment.char = '?')
stat_df <- pivot_longer(stat_df,precision:f1.score,"Metric")
ggplot(stat_df,aes(x=contig,y=value,fill=contig))+
geom_bar(stat="identity") +
facet_wrap(~Metric, ncol = 1)+
theme_bw() +
theme(
axis.ticks.x = element_blank(),
axis.text.x = element_blank()
) +
theme(legend.position="none")